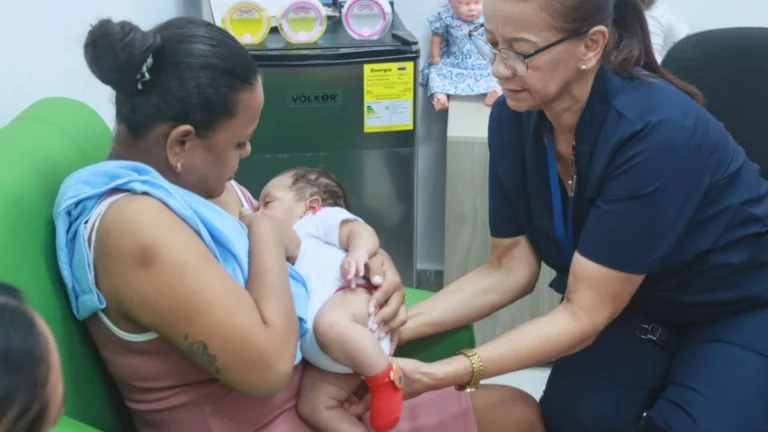
El pasado 26 de Julio de 2019 fue aprobada la ley 1980 que tiene por objeto regular y ampliar la práctica del tamizaje neonatal en Colombia mediante la detección temprana de ceguera y sordera congénitas, la utilización, almacenamiento y disposición de la muestra de sangre del recién nacido.
Ahora el Ministerio de Salud y Protección Social, deberá encausar esta norma para garantizar que los recién nacidos puedan de manera gratuita, obligatoria y progresiva, tener acceso al tamizaje neonatal básico, auditivo y visual enmarcado dentro de los lineamientos de salud pública y del modelo de prestación en redes integrales de atención en salud.
También puede leer: tamizaje neonatal pasaría a ser nueva ley de la república
Importancia de la Ley tamizaje neonatal
Con esta norma se pretende llevar acabo un conjunto de acciones como la toma de muestras de sangre del cordón umbilical o del talón a los niños recién nacidos para detectar tempranamente los errores congénitos del metabolismo y enfermedades que puedan deteriorar la calidad de vida de los bebés y dejar secuelas futuras.
Beneficios de la norma
Por consiguiente, la nueva normatividad permitirá que los aproximadamente 660.000 niños que en promedio nacen cada año en el país se beneficien de la aplicación de siete pruebas que se realizan para identificar de manera temprana igual número de enfermedades severamente discapacitantes. Son pruebas que, a partir de análisis de sangre, permiten detectar hipotiroidismo congénito, trastorno de hiperplasia suprarrenal congénita, fenilcetonuria, galactosemia, fibrosis quística, hemoglobinopatías y déficit de biotinidasa, además de buscar ceguera y sordera en los recién nacidos.
Lineamientos técnicos de las muestras
Así mismo, la ley indica que el Instituto Nacional de Salud, actuará como Centro Nacional Coordinador del Tamizaje Neonatal, a través de la Dirección de Redes, dando los lineamientos técnicos para la toma de la muestra, transporte, almacenamiento, procesamiento y entrega de esta.
Por otra parte, según la representante del proyecto de ley de tamizaje neonatal, Margarita Restrepo aseguró “esta es una gran noticia para los niños de nuestro país, estamos legislando a favor de la promoción y prevención en salud, recordemos que en el año 2015 murieron 168 niños por causa de las enfermedades que ahora serán incluidas en el tamizaje”, sostuvo la representante.

¿Qué pasará con los bebés no tamizados?
En Consecuencia, subrayó el senador ponente de la iniciativa, Antonio Correa, “el Instituto Colombiano de Bienestar Familiar (ICBF) tendrá la responsabilidad de reportar los bebés no tamizados que se encuentren dentro de los rangos requeridos para la prueba y se asegurará de coordinar el examen con la Secretaría de Salud correspondiente.
Así mismo, “en las 72 horas siguientes al nacimiento, se deberán hacer exámenes de sangre con el fin de detectar tempranamente problemas de ceguera o sordera congénitas”. aseguró Correa.