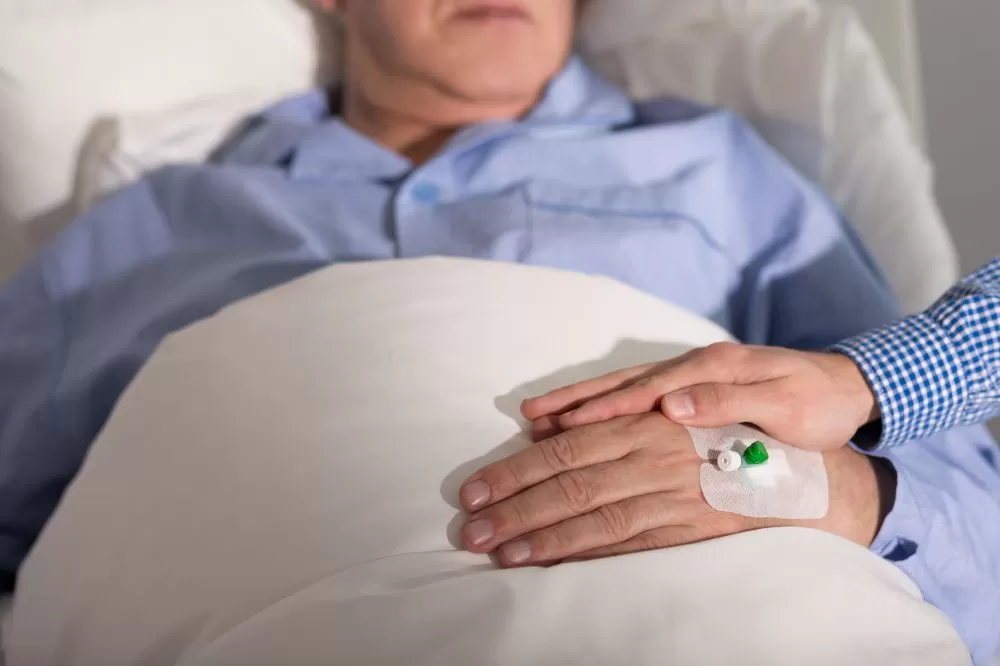
Considerando los derechos del paciente, el principio y valor de dignidad humana, el libre desarrollo de la personalidad, la autonomía, la intimidad y el derecho a no ser sometido a tratos crueles, inhumanos y degradantes, el Ministerio de Salud amplió las disposiciones del Documento de Voluntad Anticipada – DVA.
ANTECEDENTES
La muerte digna en Colombia ha logrado a través de regulaciones del Ministerio de Salud: La ley de cuidados paliativos, después llegó la regulación de la eutanasia para adultos y menores de edad, y ahora la voluntad anticipada.
¿QUÉ ES UN DVA?
Es la expresión autónoma, responsable, libre, espontánea y consciente de la voluntad de someterse o no a medios o tratamientos y procedimientos médicos innecesarios que pretendan prolongar su vida. Tiene estrecha relación con la figura del consentimiento informado.
RESOLUCIÓN 2665 DE 2018
El Ministerio de Salud expidió la resolución 2665 de 2018, que deroga la resolución 1051 de 2016 sobre el Documento de Voluntad Anticipada, ampliando sus disposiciones y reglamentando parcialmente ley 1733 de 2014, conocida como la Ley Consuelo Devis Saavedra (Nombre dado en honor a una mujer que permaneció 14 años en coma, caso por el cual se planteó la posibilidad de regular los cuidados paliativos), que regula los servicios de cuidados paliativos para enfermedades terminales, degenerativas, crónicas e irreversibles.
La anterior normativa se cerraba a pacientes que padecían enfermedades terminales, crónicas, degenerativas e irreversibles con alto impacto en la calidad de vida, a partir de ahora, se amplía su interpretación a toda persona capaz, sana o en estado de enfermedad en pleno uso de sus facultades legales y mentales, con conocimiento total de las implicaciones que acarrea este derecho, podrá suscribir el DVA.
Es decir, que no se centra solo en la decisión de ser sometido o no la eutanasia, como muchos lo interpretan, sino que hace alusión a que la persona podrá decidir el tipo de cuidados paliativos que quisieran recibir, por ejemplo si desea ser sedado o sometido a ciertos procedimientos, si desea estar acompañados por familiares, pasar sus últimos instantes en un hospital o en casa.
REQUISITOS PARA LA DECLARACIÓN:
Facultades legales y mentales, a partir de los 14 años se puede suscribir el documento. Si la persona se encuentra entre los 14 y 17 años, podrá suscribir el DVA, y deberá reemplazarlo al cumplir los 18 años por otro DVA que refleje su voluntad.
El DVA debe incluir:
-
Nombres y apellidos.
- Identificación.
-
Indicar de forma clara y específica que la manifestación se hace en pleno uso de sus facultades mentales y libre de toda coacción
-
Estar informado de las implicaciones de la declaración.
-
Manifestación clara, expresa e inequívoca respecto de sus preferencias en relación al cuidado futuro de su salud e integridad física, indicaciones concretas de su cuidado y preferencias al final de la vida.
-
Firma (de no saber firmar lo podrán hacer dos testigos o un familiar)
-
Voluntad de donación si así lo quisiera.
Este se debe autenticar ante un notario a través de escritura pública, ante dos testigos o ante el médico tratante y podrá ser modificado o sustituido en todo o parte del contenido, acudiendo a las mismas vías.
El DVA se debe anexar a la historia clínica y ser cumplido en su totalidad, el personal que administre o conozca de su contenido deberá guardar reserva de esta información.
Adicionalmente el Ministerio de Salud abre la puerta para que sirva como registro de la voluntad anticipada del paciente, medios tecnológicos y lenguajes alternativos.
A través de la página web del Ministerio de Salud, se dispondrá el formato de DVA.
El DVA protege en todo momento la dignidad de la persona y garantiza el cumplimiento de su voluntad y su derecho al libre desarrollo de la personalidad y a la autonomía.
Descargue la Resolución 2665 y conozca todos los detalles.





















